Importing sequences¶
You can load sequences into OpenCloning from many places. You can either drag-and-drop files, or use the import sequence form. If you want to load more than one sequence via sequence form, you can add more "lanes" by clicking on the + button.
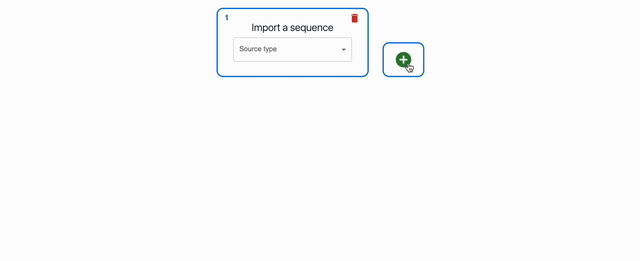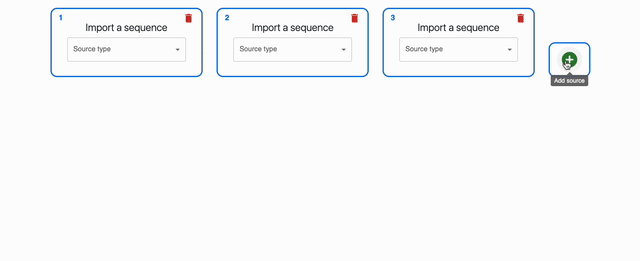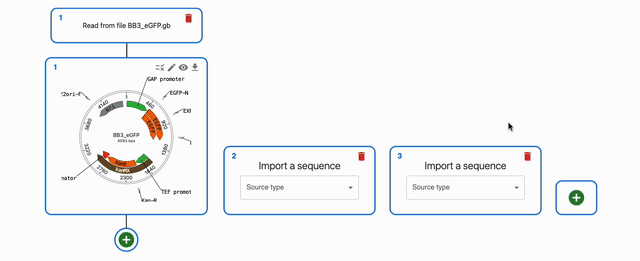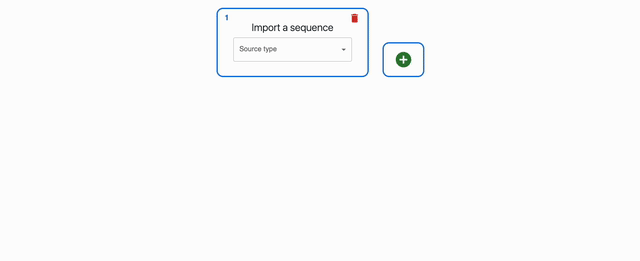
Loading your own files¶
Drag-and-drop¶
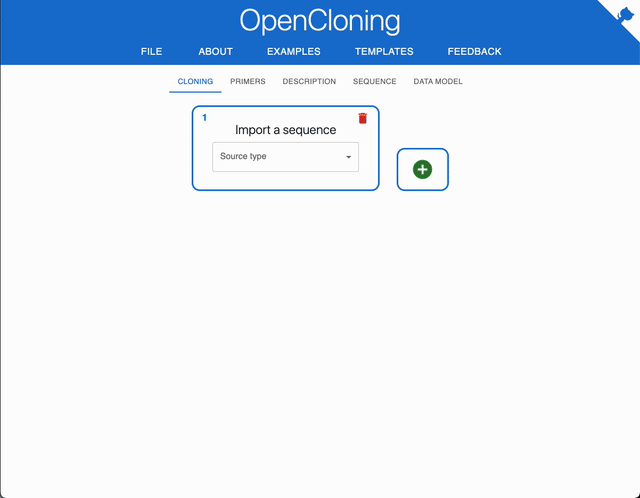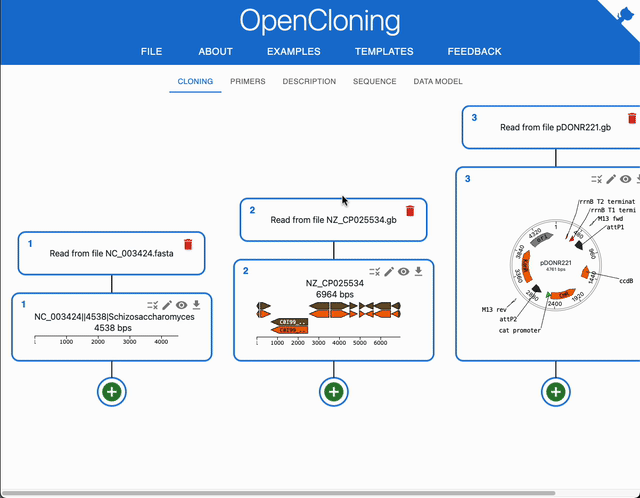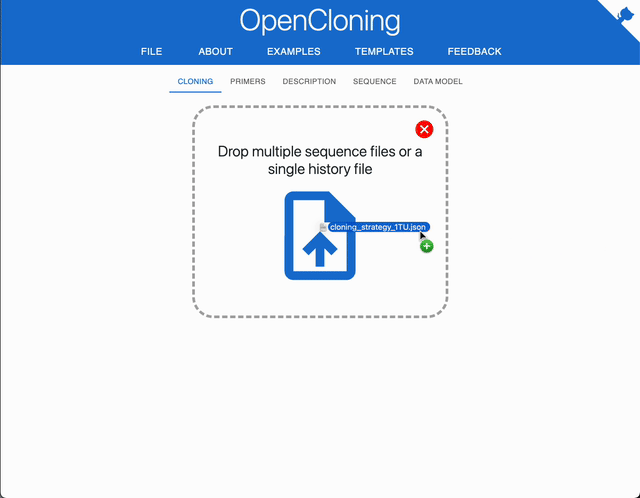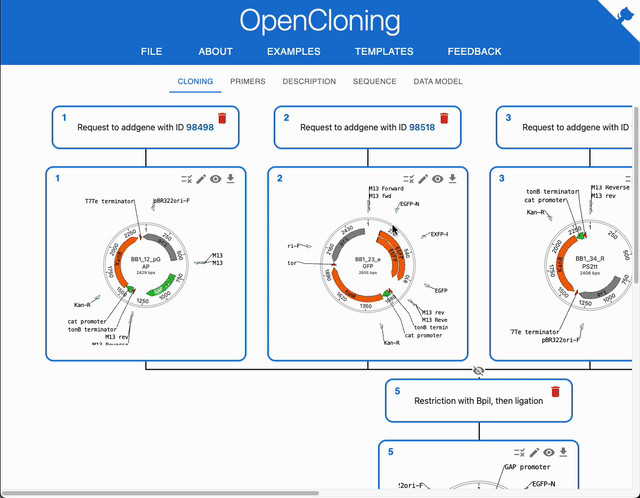
The easiest way to load your files is to drag and drop your files into the application. You can drop:
- Multiple sequence files: FASTA (
.fasta,.fa), GenBank (.gb,.gbk), SnapGene (.dna), ApE (.ape).⚠️ If a FASTA or GenBank file dropped contains multiple sequences, only the first one will be loaded.
- A single
.jsonor.zipfile containing a cloning strategy (see exporting docs).
Use "Submit file"¶
You can load an individual file, by selecting "Submit file" in "Source type". This allows some extra options:
- If you tick the option
Circularize, the sequence will be circularized. This can be useful if you are loading a plasmid from a FASTA file, where topology is not specified, or if you have a genbank file with a plasmid with wrong topology in the metadata. - If you tick the option
Extract subsequence, you can load a part of the sequence provided in the file by indicating the start and end positions. This is particularly useful if you are using a file that contains a large sequence, such as a chromosome. - If the file contains multiple sequences, you will be able to select which one to load.
Use "File > Load cloning history from file"¶
If you want to load a .json or .zip file containing a cloning strategy, you can use the "File > Load cloning history from file" menu item.
Loading sequences from a repository¶
OpenCloning can load sequences from multiple repositories. On Source type, select Repository, and choose from the list of available repositories.
- Some, like Addgene, will require you to provide an identifier. For Addgene, you can find that in the url of the plasmid page, for instance, in
https://www.addgene.org/39292/, the id is39292. - Others, like SEVA plasmids, will have a drop-down menu to select from options.
Loading genome sequences from NCBI¶
On Source type, select Genome Region, which enables several options:
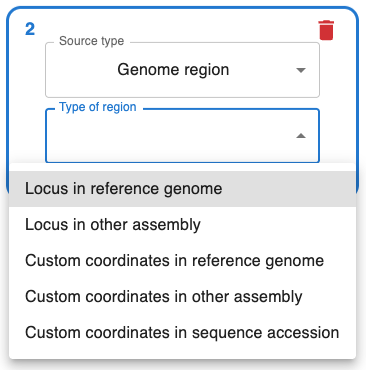
- When using a reference genome, you can query by species and the reference genome assembly for that species will be used.
- If you are working with an assembly other than the reference genome, select
other assemblyand provide the assembly accession. To find your accession number, I recommend using the NCBI datasets Genome page. You can type your species / taxon, and you will find all associated assemblies. - If you select
Locus, you will be able to use the genome annotation to find a locus of interest, for example querying by gene name. This relies on the annotation of the genome in the NCBI. If you can't find something, see if you can find it in the NCBI datasets Genome Annotation page for that assembly. For example, if you are working with assemblyGCA_000002945.3, go to https://www.ncbi.nlm.nih.gov/datasets/gene/GCA_000002945.3/. - If you select
Custom coordinates, you will have to choose the chromosome or contig within the assembly, then select the start and end, and the strand (selectminusif you want the reverse complement). - If you want to use a sequence that is not part of an assembly (e.g. a viral genome), you can select
Custom coordinates in sequence accession. Unfortunately so far you cannot query by annotation with this option.
Problems with annotation?
If you can't find the annotation you expect in the assembly, it may be that the annotation is different in the RefSeq or GenBank genome 🔍. This is rare, but it can happen!
For example, the GenBank genome GCA_000744395.1 does not have annotations, but its equivalent RefSeq genome GCF_000711245.1 does ✨. In a case like this, OpenCloning will give you a warning and suggest to use the RefSeq genome instead.
However, RefSeq and NCBI assemblies may have different annotations, and in that case you won't get a warning. If you want to find the equivalent RefSeq / NCBI, use the NCBI datasets page by replacing the assembly accession in the url https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_000711245/. There, you will see both accessions! 🎯
Manually adding sequences¶
On Source type, select Enter manually, and you will be able to add a sequence as text. It must only contain ACGT characters. You can choose whether the sequence is circular, and specify overhangs. Overhangs can contain positive and negative values. The following settings:
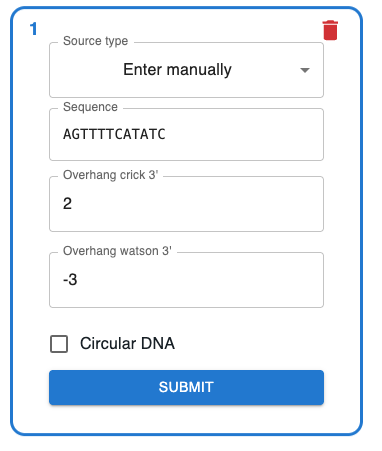
Would produce the sequence below. For full documentation on the meaning of overhangs, check the meaning of crick_ovhg and watson_ovhg in the pydna documentation.
ttttcat
|||||||
tcaaaagtatag
Loading sequences from an Electronic Lab Notebook¶
If you are using eLabFTW, you can load and save sequences to your electronic lab notebook. To see how it works, check the eLabFTW integration documentation.
Loading sequences stored as static files in the server¶
If you are running your own instance of OpenCloning (see running locally), you can mount a directory into the docker container that contains:
* An index.json file with a directory of sequences and syntaxes in your repository.
* The sequences themselves as fasta, genbank, snapgene or ape files.
To see how to set this up, check the docker-compose.yml file (STATIC_CONTENT_PATH env var and volumes). For an example of directory structure and index.json file, see the example directory.
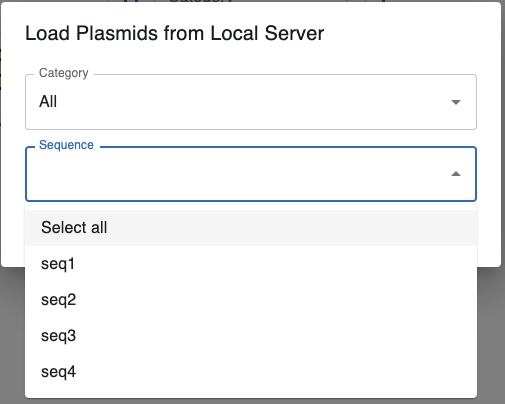
If you set this up properly, in the Source type dropdown when importing a sequence, you should see the option Local server file. Sequences can be associated with categories in the index.json file, and should be able to filter by category when selecting a sequence.
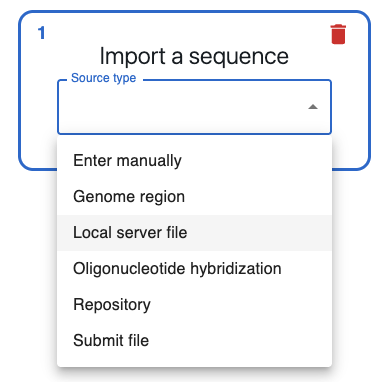
You should also be able to import a syntax from the server in the Assembler tab when you click on Load Syntax.
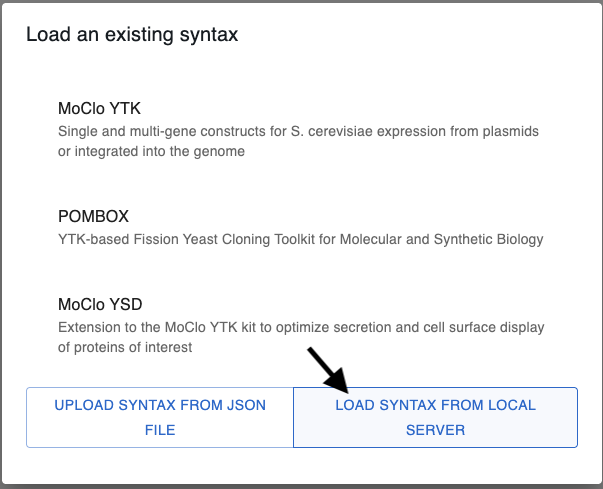
Finally, when the syntax is loaded, you should be able to import multiple sequences at once in the Assembler tab:

Clicking there, you will be able to select multiple sequences to import at once. You can import all sequences within the chosen category if you select Select All in the Sequence dropdown.